
Good Design Controls Are Critical to Avoid FDA Issues
We recently sat down with a few medical device companies to discuss how they deal with FDA requirements around design controls. Medical device companies recognize that having an emphasis on design controls helps to assure good design and engineering practices that yield high-quality solutions. But to accomplish this, first know what key elements should be incorporated in design controls before implementing formalized plans for their creation.
In addition to achieving better practices and designs, why should medical device companies focus on design controls? Establishing sound design controls can prevent FDA inspection observations and —which can be very costly and even derail product launches. Both Form 483s and FDA warning letters are similar in nature and serve to inform all affected parties regarding issues that require corrective action. The Form 483s, also called “Inspectional Observations,” convey conditions or practices that indicate a potential violation of FDA requirements. The warning letter is typically an escalation from insufficient follow-up on a 483.
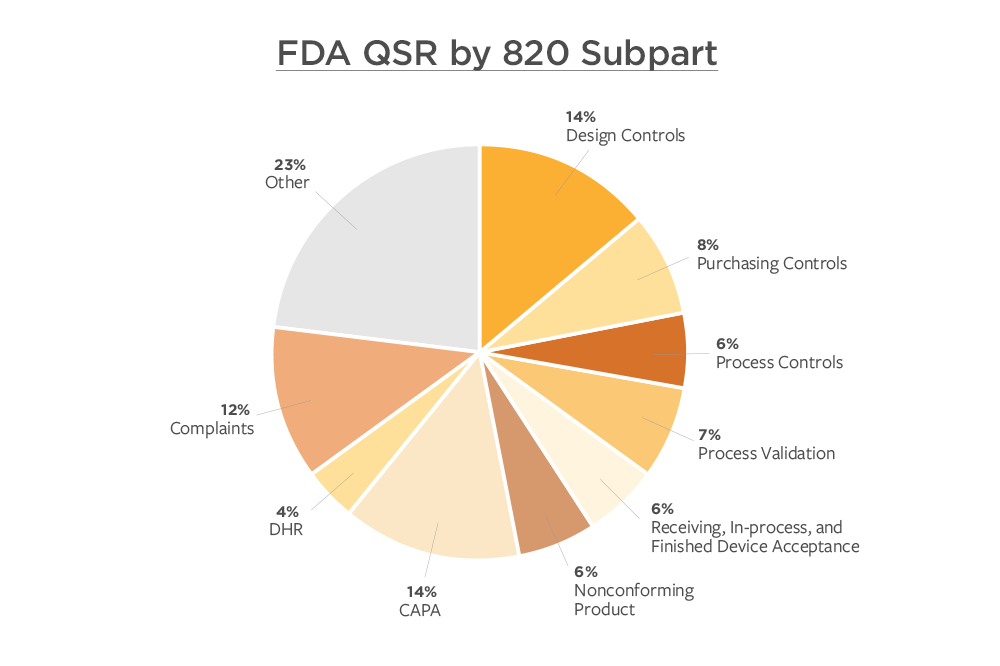
Approximately 90% of 2018 FDA 483s cite Quality System Regulation issues (21 CFR 820), which are very concerning. The preceding pie chart shows that 14% are design control issues (CFR 820.30) as well as corrective and preventive actions (CAPA). The consequences are significant to affected companies. Medical device manufacturers avoid being listed on the FDA website, as this kind of visibility can be detrimental to future sales and profitability.
- Patients and physicians may lose confidence.
- Competitors can exploit the information.
- Management and other resources can be pulled from priorities to address issues.
Key Elements of Design Controls
A solid design controls foundation simplifies FDA compliance, so you pass audits without a finding. These are the key elements of design controls.
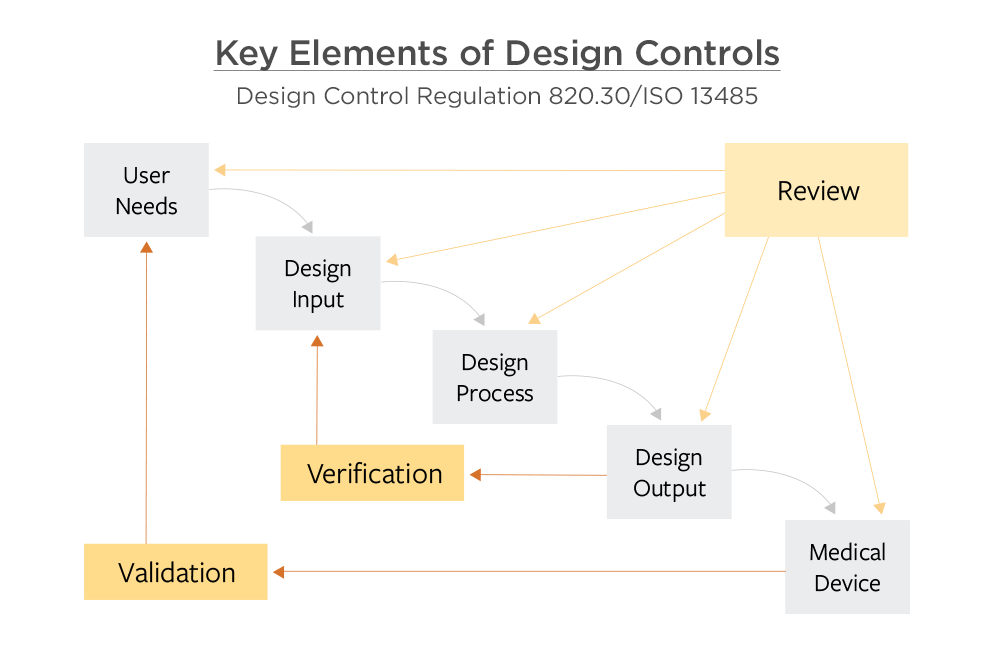
Design and development planning is the first element in the design control process. The plan includes activities, dates, and responsible people. The plan is documented and updated as it evolves. This leads to the next element, design input. Design input comes from users, patients, and physicians along with other stakeholders. The design process results in design output, which is documentation describing the product. Design reviews are achieved throughout design development in order to check the design results and identify issues.
Verification ensures that the design outputs completely and unambiguously match the design input. During verification, you should be able to answer questions like:
- “Do we have all the documentation we need?”
- “Who has approved it and when?”
- “What’s missing?”
Design transfer is the process of providing manufacturing with the design output they need, including BOMs, drawings, and assembly instructions. Design transfer is not frequently cited in 483s, yet we frequently see issues with companies’ design transfer processes. Often companies transfer information manually from engineering to operations, resulting in greater risk of delays and errors.
The next element is validation—testing to make sure the manufactured product meets the users’ needs. All these tests and results must be documented. Validation is the most commonly referenced sub-part of the design controls 483s.
Design changes are the second-most cited issue under design controls. After releasing the first revision, updating ERP and other downstream systems can be tricky. Many aspects of the design may have changed, which makes it essential to understand:
- What’s changed?
- What components were added or removed?
- Has all the supporting documentation been updated?
Change processes can have many variables making documenting and following those processes more difficult.
Finally, the design history file (DHF) is the compilation of a company’s product design documentation and its evolution. This is an administration-heavy element that can be cumbersome to maintain and use.
Good Design Controls Are Critical to Avoid FDA Issues
Traditional, document-centric QMS solutions can help simplify some of these processes. However, a product-centric QMS solution leverages a single system to manage the foundational product record in context with all related quality processes. This connected product record includes a relational bill of materials (BOM), approved manufacturers list (AML), and changes (ECOs). Managing the quality processes in the same system creates better visibility of issues and faster resolution with connected quality records like complaints and corrective and preventive actions (CAPAs) that can drive ECOs to correct any design issues.
Ultimately, having a product-centric QMS helps companies with design transfer and design change processes as well as the FDA required file management. This also helps medical device companies establish solid design controls to avoid FDA warnings that can result in lost revenue, patient health issues, or, in some cases, death.
Medical device companies need full control, visibility, and traceability throughout the design, development, and manufacturing process.

